Usage¶
This is a general description of the usage and function of each tool found in the VirAmp pipeline. A more detailed description can be found at the webpage of each tool.
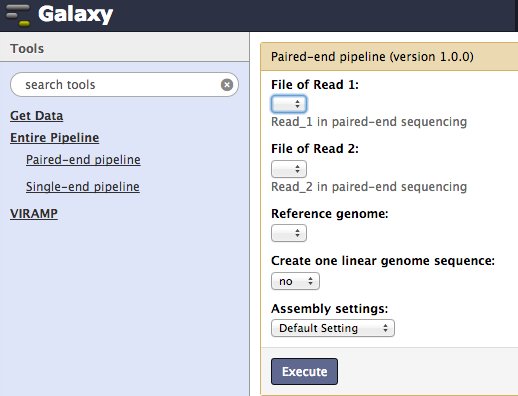
One-click pipeline¶
Two general pipelines are provided with a one-click option, one for paired-end data and the other for single-end data. Users are only required to submit read files and a reference file corresponding to their data. Alongside the default settings, users may use the “advanced setting” option to custom configure the pipeline with alternative parameters.
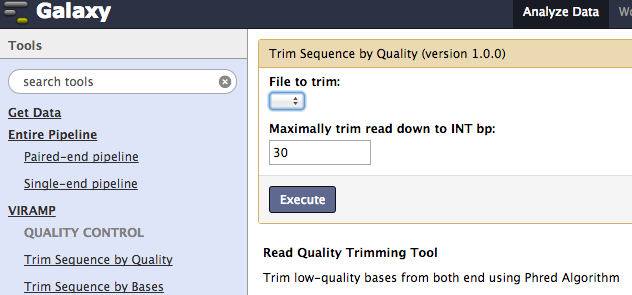
Quality Control¶
First, trim out the low quality bases of the input fastq files. This can be achieved by either removing low quality bases or trimming a certain length from each end.
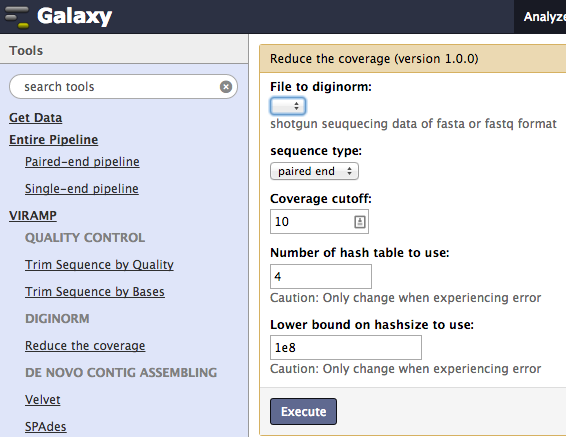
Diginorm¶
Next, reduce coverage and bias using Digital normalization. This step reduces the sample variation as well as sample bias.
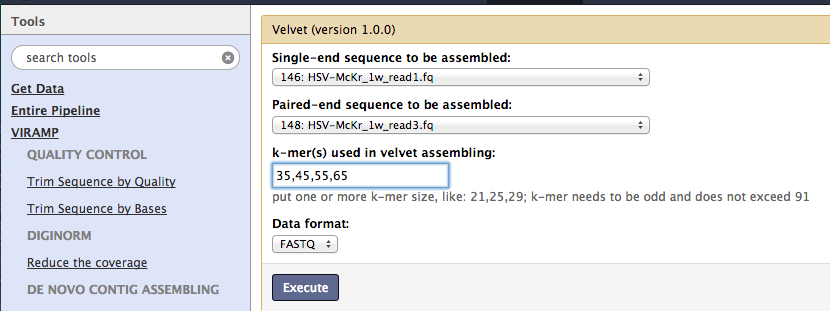
de novo Contig assembly¶
Now, the pipeline assembles the short reads into longer contigs. By default the One-click pipeline uses velvet. Two alternatives, SPAdes and VICUNA , are provided and can be selected as either individual tools or through the advanced options in the one-click pipeline.
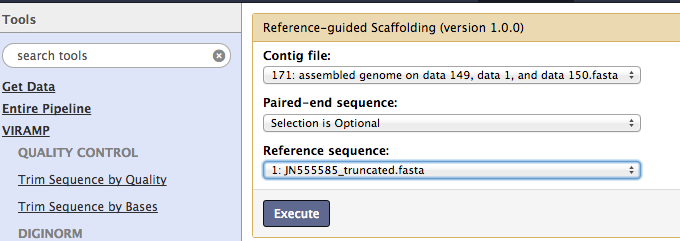
Reference-based scaffolding¶
The contigs are then assembled into even longer super-contigs. This step is a modification of AMOScmp
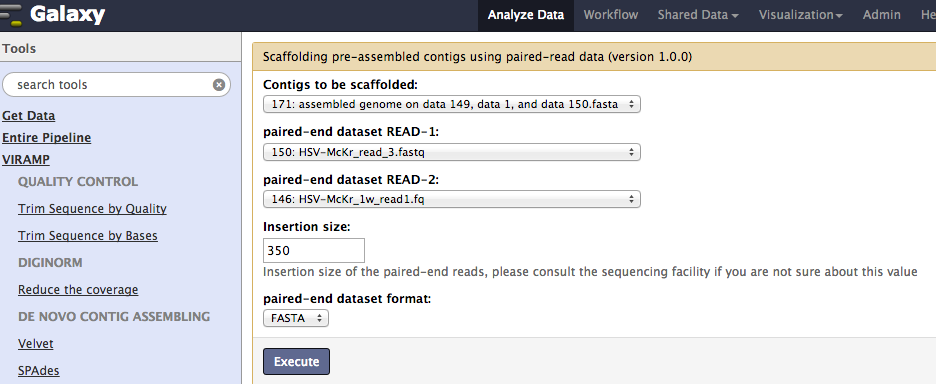
Reference-independent scaffolding¶
The next step extends the super-contigs and connects them using SSPACE. The pipeline will produce a draft genome as a multi-fasta file usually containing 5~15 contigs which are listed in the same order as the reference.
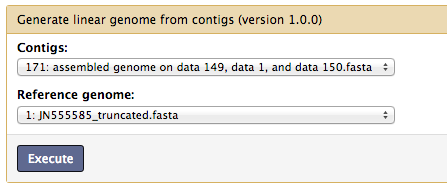
Gap closing¶
This step connects all the contigs in the multi-fasta from the previous step into one linear genome for the convenience of downstream functional analysis. However, this is optional and highly recommended to be done only after assessing the draft genome, as the gaps between the contigs could be from misassembly, sequencing, genome feature, etc.